Learning Circos
Bioinformatics and Genome Analysis
Institut Pasteur Tunis Tunisia, December 10 – December 11, 2018
v2.00 6 Dec 2018 Download PDF slides
v2.00 6 Dec 2018
A 2- or 4-day practical mini-course in Circos, command-line parsing and scripting. This material is part of the Bioinformatics and Genome Analysis course held at the Institut Pasteur Tunis.
Quick links
BCGA 2018 | 1-day Circos course | Circos documentation best practices getting started | Brewer palette swatches | Color resources | Nature Methods Points of View Points of Significance
Circos case studies
Tuesday 11 December 2018 — Day 2
9h00 - 10h30 | Lecture 1 — Drawing the human genome
11h00 - 12h30 | Lecture (practical) 2 — Downloading and drawing human genes
14h00 - 15h30 | Lecture (practical) 3 — Downloading and drawing segmental duplications
16h00 - 18h00 | Lecture (practical) 4 — Creating an image montage
Concepts covered
drawing the human genome, karyotypes included in Circos distribution, generating random data with randomdata, heatmaps, color lists, interpolating colors with colorinterpolate, drawing human genes, drawing segmental duplications as links, creating image montages
Downloading and drawing segmental duplications
In this lecture, you'll draw human segmental duplications as links. You'll find answers to this challenge in Lecture 4.
Download segmental duplications
You can get the human segmental duplication set from the UCSC genome browser table viewer.
clade: Mammal
genome: Human
assembly: Dec 2013 (hg38)
group: Repeats
track: Segmental Dups
table: genomicSuperDups
To learn about the format of this file, click on "Describe table schema".
There are about 70,000 these duplications, which are all at least 1 kb.
I encourage you to download the file yourself but if the network connection is slow, you can find it in day.2/data/segdup.human.txt.
Create a link file
Parse the file to generate a link file based on the coordinates of the two duplicated regions in each line.
Generate a histogram of the sizes of the segmental duplications. You can use the size on the first chromosome. What is the average size?
Create a bash script to list the largest N segmental duplications on each chromosome. For this you want to use a bash for loop.
https://unix.stackexchange.com/questions/209249/piping-commands-after-a-piped-xargs
Now use this script to modify your link file to add an sizerank=IDX column to the link file where IDX is the size rank of the duplication on its chromosome.
hs1 146541435 146905930 hs16 70811383 71168670 sizerank=1
hs1 148600078 148935345 hs1 119989247 120323081 sizerank=2
hs1 119989247 120323081 hs1 148600078 148935345 sizerank=3
...
hs2 110276210 110634615 hs2 109736854 110095177 sizerank=1
hs2 109736854 110095177 hs2 110276210 110634615 sizerank=2
hs2 94571013 94860516 hs9 65858856 66156287 sizerank=3
...
The final output file should be sorted by link size (not the rank) and should look something like this
hsY 5464146 6234575 hsX 92352303 93120510 sizerank=1
hsX 92352303 93120510 hsY 5464146 6234575 sizerank=1
hsY 25545548 26311622 hsY 23358995 24124586 sizerank=2
hsY 23358995 24124586 hsY 25545548 26311622 sizerank=3
hsY 24894109 25541603 hsY 24128531 24775999 sizerank=4
hsY 24128531 24775999 hsY 24894109 25541603 sizerank=5
hsX 90276317 90909509 hsY 3853083 4483712 sizerank=2
...
Draw the segmental duplications
Create a <link> track in the template etc/ Circos configuration file to draw the segmental duplications.
First, start by drawing the largest 100 links on each chromosome. Since you've stored their rank in sizerank you can test in a rule
<rule>
condition = var(sizerank) > 100
show = no
</rule>
##3 Color segmental duplications by chromosome
Add a rule that colors the links by their chromosome. What happens here if you use chr2?
<rule>
condition = 1
color = eval(lc var(chr1))
</rule>
To this same rule and a line that set's the link's z parameter to the same as its rank. This parameter controls the order in which the links are drawn, in order of decreasing z. Thus, links with sizerank=1 are drawn last.
z = eval(var(sizerank))
Add a rule that draws links with sizerank<10 with thickness=2.
<rule>
condition = var(sizerank) < 10
thickness = 3
</rule>
Map size onto color
Apply a color map to map size1 of the duplication onto the spectral-9 Brewer palette. For this what you want to do is remap the var(size1) of a link from the range, for example, [1000,20000] to the colors spectral-9-div-1 to spectral-9-div-9.
color = eval(sprintf("spectral-9-div-%d",remap_int(var(size1),1000,20000,1,9)))
Try other color palettes like piyg and reds. Remember that piyg is a diverging Brewer palette so its name has -div whereas reds is sequential so it has -seq.
You'll be working towards images like the ones below.
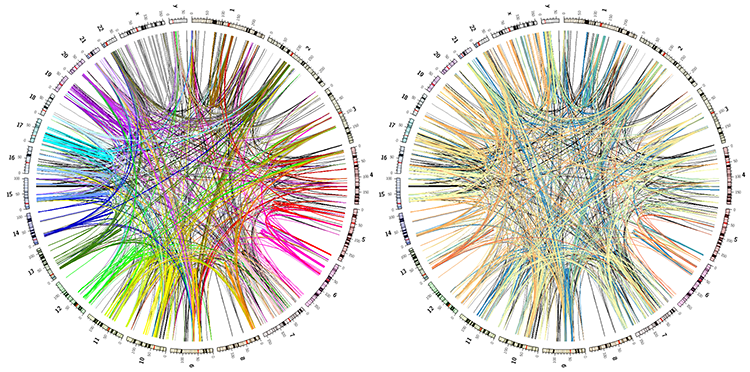
karyotype = data/karyotype/karyotype.human.txt
This parameter is normally set to 25,000 in etc/housekeeping but here we will override it since we might be drawing more than 25,000 links. Keep in mind that drawing that many links can create an uninterpretable image and take a long time to draw.
max_links* = 100000
#<links>
#<link>
For debugging, you can set record_limit to read in a small number of entries from the track data file.
#record_limit = 5000
#</link>
#</links>
