Learning Circos
Bioinformatics and Genome Analysis
Institut Pasteur Tunis Tunisia, December 10 – December 11, 2018
v2.00 6 Dec 2018 Download PDF slides
v2.00 6 Dec 2018
A 2- or 4-day practical mini-course in Circos, command-line parsing and scripting. This material is part of the Bioinformatics and Genome Analysis course held at the Institut Pasteur Tunis.
Quick links
BCGA 2018 | 1-day Circos course | Circos documentation best practices getting started | Brewer palette swatches | Color resources | Nature Methods Points of View Points of Significance
Getting started with Circos: Yeast and Ebola
Monday 10 December 2018 — Day 1
9h00 - 10h30 | Lecture 1 — Introduction to Circos
11h00 - 12h30 | Lecture (practical) 2 — Visualizing gene distribution and size in Yeast: the histogram data track
14h00 - 15h30 | Lecture (practical) 3 — Conservation in Yeast: the link data track
16h00 - 18h00 | Lecture (practical) 4 — Visualizing an Ebola strain
Concepts covered today
Circos configuration, common Circos errors, Circos debugging, ideograms, selecting ideograms with regular expressions, data tracks, histograms, links, downloading files from UCSC genome browser, essential command-line tools and basic scripting, using bash to create data files for Ebola genome strains, color definitions, using transparency, Brewer palettes, runtime formatting rules, accessing data track statistics, input data formats
Conservation in Yeast: the link data track
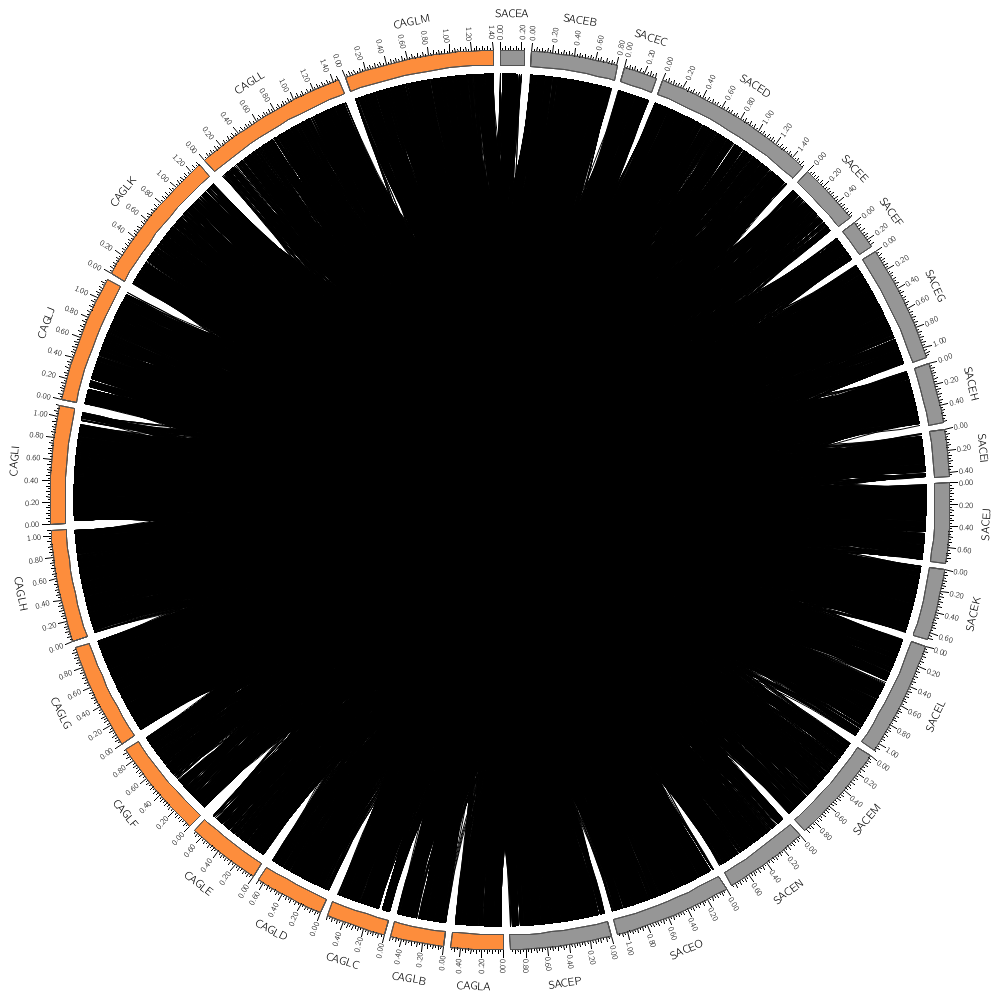
Let's now look at how to draw links. These are useful for showing alignments and any other kind of similarity (or more generally, relationship) between two positions.
Let's draw chromosomes from a couple of the genomes, SACE and CAGL.
Here the regular expression is using the alternation pipe, meaning that chromosome names can match either "sace" or "cagl"
chromosomes_display_default = no
chromosomes = /sace|cagl/
Like plots, links are defined in blocks. Unlike plots, they belong in <links> blocks. Each link track has its own <link> block.
<links>
<link>
file = ../../data/link_cagl_sace.txt
Different block types expect different parameters. For the links, you need to define the position of the ends of the links. This is best done relative to the ideogram of the circle.
So here our links will go as far as 98% of the radius of the ideogram circle.
radius = 0.98r
This controls how curved the links are. Specifically, this is the radius of the control point. Try setting this to a larger value like 0.25r or 0.5r.
bezier_radius = 0r
Since there are about 10,000 links in the file, the image might take a while to draw. The record_limit parameter is useful for limiting the number of entries read from the file for debugging. Once you're happy, comment out this line to draw all the links, which may take 20-30 seconds.
#record_limit = 1000
color = black
thickness = 1
</link>
</links>
Since we'll always be using this karyotype file for today, for the rest of today's lectures this line has been moved to the bottom of the file so that the things at the top are the things we're focusing on.
karyotype = ../../data/karyotype.txt
chromosomes_display_default = no
chromosomes = /sace|cagl/
<links>
<link>
file = ../../data/link_cagl_sace.txt
radius = 0.98r
bezier_radius = 0r
thickness = 1
#record_limit = 1000
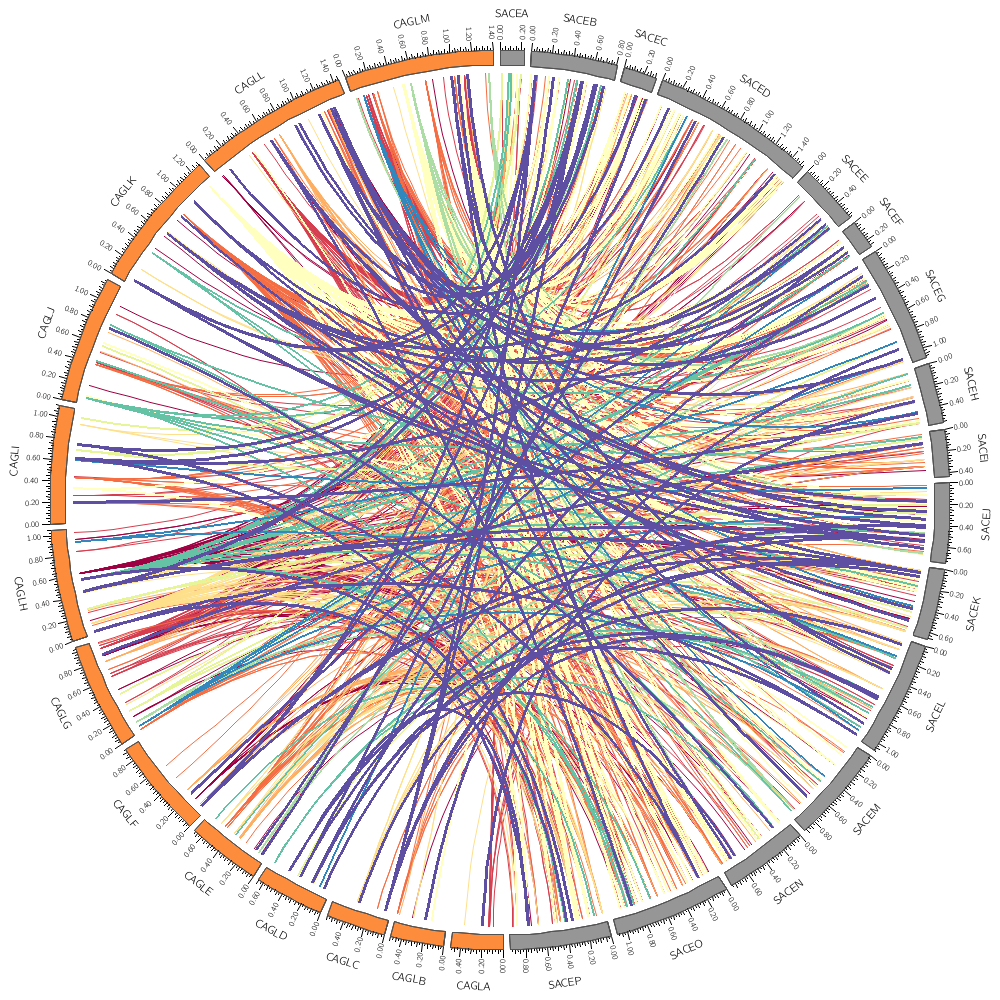
You'll notice in the previous part that all the links were black and because there were so many of them, they overlapped and the image was saturated with black.
You can set a color's transparency by using the _aN suffix on the color's name. In the underlying Circos configuration files (which are included at the bottom of this file) I've defined a parameter
auto_alpha_steps = 20
This gives you 20 levels of transparency for each color.
black -> opaque
black_a1 -> 1/21 transparent
black_a2 -> 2/21 transparent
...
black_a20 -> 20/21 transparent
For obvious reasons, you don't need a 100% transparent version of each color, so the denominator in the fractions above is N+1. If you're interested look in the sessions/day.1/etc directory. If you follow all the include directives up the tree, you'll find that this file is included by all the parts of all the lectures for this day.
color = black_a15
Even with _a15 (75% transparency) the image looks quite busy. Try _a20. Now we come to a very powerful part of Circos. Instead of just drawing the data the way it appers in the input file, you can adjust how data points (e.g. histogram bins, links, or anything) can appear based on its properties, such as chromosome, position, size, and so on.
While you can potentially define these parameter in the input files, by applying them to only certain lines in the files that you filtered with a script, it's easier to do it in the configuration file.
Turn the rules on below by setting use=yes.
<rules>
use = yes
One of the simplest ways in which a rule can be used is to turn of the display of data points.
Since our links are regions of conservation, let's only show those that have a size of at least 4 kb. To do this, we set a condition that triggers for links smaller than 4 kb. Any other directives or parameters in the rule will be applied to these links before they're drawn. In this case setting show=no will hide the links.
<rule>
condition = var(size1) < 4000
show = no
</rule>
You can change the color of a data point (which may be a point in a scatter plot# bar in a histogram, a link, etc) using any of its other property. Each link here corresponds to a conserved region in CAGL that also appears in SACE and thus the start and end of the link have a size.
The rule below maps the color of the link based on the size of its start given by var(size1) onto the 11-color diverging spectral Brewer palette. Two additional lines change the thickness of the link and its z-depth.
Circos defines a large number of named colors. All these definitions are in the Circos installation directory under etc/. For convenience, I have included these files in colornames/ in this lecture's section.
<rule>
use = yes
This condition applies to all data points that reach this rule.
condition = 1
The function remap_int(var,min,max,mapmin,mapmax) linearly maps the value given by var from the range [min,max] to [mapmin,mapmax]. We're using sprintf(fmt,list) to format the value as a string that corresponds to the name of a Brewer color.
The eval() is required to parse the contents as code as opposed to treating them as a string literal.
color = eval(sprintf("spectral-11-div-%d",remap_int(var(size1),4000,6000,1,11)))
thickness = eval(sprintf("%d",remap_int(var(size1),4000,6000,1,3)))
Without eval(), z would be set to the literal string "var(size1") and not to the actual value of the size1 parameter.
z = eval(var(size1))
</rule>
</rules>
</link>
</links>
karyotype = ../../data/karyotype.txt
Below, I show you a part of the Circos configuration file that defines the Brewer palette colors. You can find this file in etc/colors.brewer.conf in the Circos installation directory.
Colors from www.colorbrewer.org by Cynthia A. Brewer,
Geography, Pennsylvania State University. See BREWER for license.
Color names are palette-numcolors-type-idx (e.g. reds-3-seq-1)
palette palette name (e.g. reds)
numcolors number of colors in the palette (e.g. 3)
type palette type (div, seq, qual)
idx color index within the palette (e.g. 1)
Another version of the color index is defined where colorcode is the color's letter unique to a given palette.
For each palette, two color list are defined for use with heatmaps.
palette-numcolors-type
palette-numcolors-type-rev
where the second contains colors in reversed order.
Also supported is the format
palette-numcolors
palette-numcolors-rev
(without the -type), which isn't necessary to uniquely identify the palette.
Each diverging and sequential palette has all the colors used for its n-color variants listed an integrated 13-color (for sequential) or 15-color (for diverging) palettes.
http://www.personal.psu.edu/cab38/ColorBrewer/ColorBrewer_updates.html
http://mkweb.bcgsc.ca/brewer
For example, the 3-color sequential blues palette is defined by these colors
blues-3-seq = blues-3-seq-(\d+)
blues-3-seq-rev = rev(blues-3-seq-(\d+))
blues-3-seq-1 = 222,235,247
blues-3-seq-2 = 158,202,225
blues-3-seq-3 = 49,130,189
And the 4-color version of it is
blues-4-seq = blues-4-seq-(\d+)
blues-4-seq-rev = rev(blues-4-seq-(\d+))
blues-4-seq-1 = 239,243,255
blues-4-seq-2 = 189,215,231
blues-4-seq-3 = 107,174,214
blues-4-seq-4 = 33,113,181
Here is the 11-color spectral palette
spectral-11-div = spectral-11-div-(\d+)
spectral-11-div-rev = rev(spectral-11-div-(\d+))
spectral-11-div-1 = 158,1,66
spectral-11-div-2 = 213,62,79
spectral-11-div-3 = 244,109,67
spectral-11-div-4 = 253,174,97
spectral-11-div-5 = 254,224,139
spectral-11-div-6 = 255,255,191
spectral-11-div-7 = 230,245,152
spectral-11-div-8 = 171,221,164
spectral-11-div-9 = 102,194,165
spectral-11-div-10 = 50,136,189
spectral-11-div-11 = 94,79,162
spectral-15-div-11 = 153,213,148
spectral-11 = spectral-11-div-(\d+)
spectral-11-rev = rev(spectral-11-div-(\d+))
Some tracks such as the heat maps, which map values to colors automatically, take the name of a color list, in which case you would put the name of the palette without color index
color = spectral-11-div
In most cases though you'll be refering to specific colors
color = spectral-11-div-7
chromosomes_display_default = no
chromosomes = /sace|cagl/
<links>
<link>
file = ../../data/link_cagl_sace.txt
radius = 0.80r
bezier_radius = 0r
thickness = 1
record_limit = 1000
color = black_a15
<rules>
<rule>
condition = var(size1) < 4000
show = no
</rule>
<rule>
condition = 1
color = eval(sprintf("spectral-11-div-%d",remap_int(var(size1),4000,6000,1,11)))
thickness = eval(sprintf("%d",remap_int(var(size1),4000,6000,1,3)))
z = eval(var(size1))
</rule>
</rules>
</link>
</links>
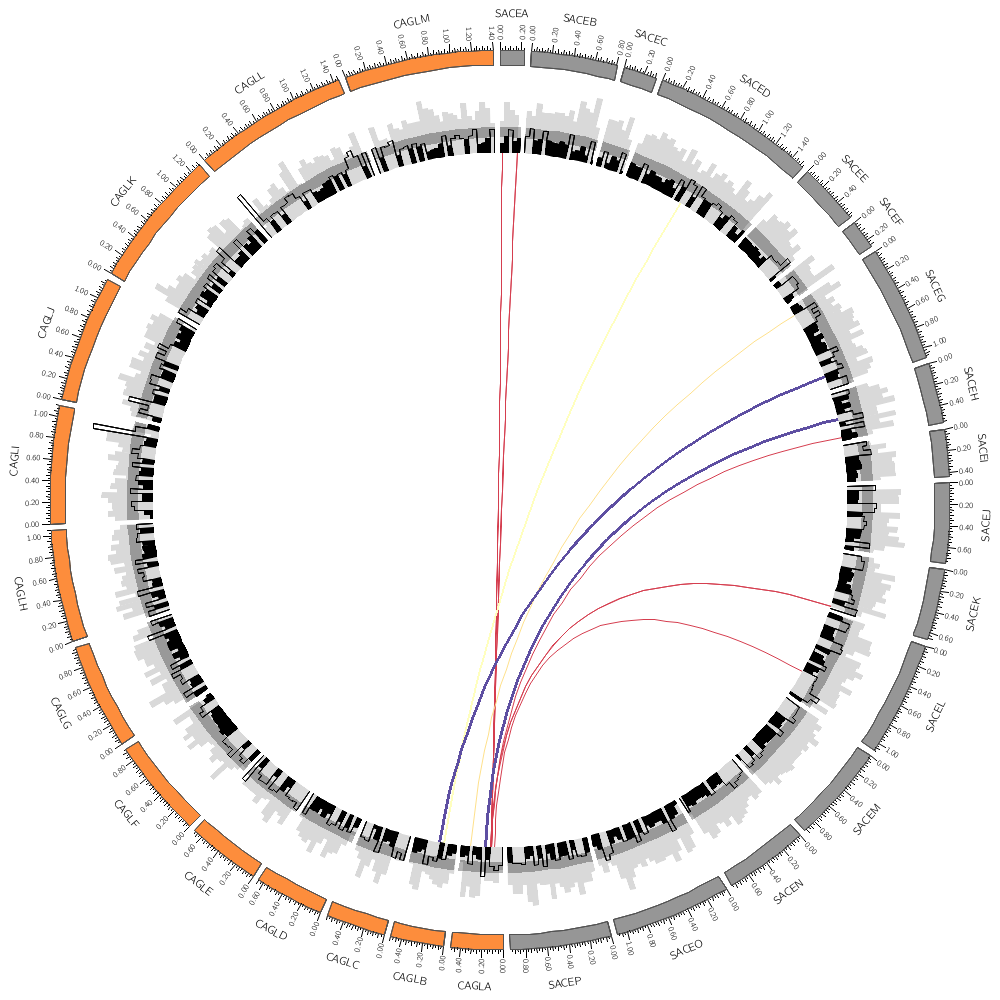
Below are the two histograms from the previous lecture. I've used the 50kb binning for both.
Notice that both tracks have the same r0 and r1 values. They are drawn on top of each other. There is no restriction on this.
<plots>
<plot>
file = ../../lecture.2/5/genes.count.50kb.txt
type = histogram
fill_color = vlgrey
stroke_thickness = 0
r1 = 0.95r
r0 = 0.80r
orientation = out
</plot>
This histogram has no fill (by default it's not defined) and a black outline.
<plot>
file = ../../lecture.2/5/genes.avgsize.50kb.txt
type = histogram
color = black
stroke_thickness = 1
r1 = 0.95r
r0 = 0.80r
orientation = out
You can reference the min, max, average (avg), standard deviation (sd) of a track using var(plot_STATISTIC). For example, var(plot_avg) gives the average and the rule above, when activated, fills assigns a black fill to bins whose size smaller than track average.
<rules>
use = no
<rule>
condition = var(value) < var(plot_avg)
fill_color = black
</rule>
</rules>
<backgrounds>
<background>
color = black_a15
y1 = 2000
y0 = 1500
</background>
</backgrounds>
</plot>
</plots>
karyotype = ../../data/karyotype.txt
Input data format
Let's look in more detail at the data input files. Their format is relatively straightforward.
First, there are only two kinds of input data files. The first kind is used by (x,y)-style tracks such as line plots, histograms, heatmaps, tiles and so on. These tracks assign a value to a region on a chromosome. The format for this is
CHR START END VALUE {options}
For example, our histogram of average gene sizes in 10 kb bins looks like this
# day1/data/genes.avgsize.10kb.txt
cagl-a 10000 19999 840
cagl-a 20000 29999 1661.4
cagl-a 30000 39999 1652.4
cagl-a 40000 49999 1828
...
If you use this file for a histogram track, the bins will be horizontal bars whose width is defined by the start and end coordinates. If you use this file for a scatter plot, then the point will be placed at the midpoint of the interval. Generally, the fourth column is the value that's used by the track and mapped onto a property, such as bar height.
It's important to stress here that the same data file can be used for various track types or reused for multiple tracks.
We'll worry about the {options} column later. This column can store additional property of the data point, such as a formatting property (color, thickness and so on, which may easily be adjusted by rules) or additional values that might be used for filtering in the track.
The second type of input file stores pairwise relationships between positions. Link files are of this form and have the format
CHR1 START1 END1 CHR2 START2 END2 {options}
For example, the conserved regions between SACE and CAGL used for the link track in this lecture are stored like this
# day1/data/link_cagl_sace.txt
cagl-a 19487 21757 sace-g 450197 452104
cagl-a 22260 24440 sace-g 452404 454560
cagl-a 25241 27622 sace-b 207194 209224
cagl-a 25241 27622 sace-g 454785 457067
...
Creating a Circos figure generally begins with filtering your data set for the kinds of things you want to display and then generating the data files from them. We'll do this in the last lecture today.