





data visualization + art
Genes that make us sick
It is said that for money you can have everything, but you cannot. You can buy food, but not appetite; medicine, but not health; knowledge, but not wisdom; glitter, but not beauty; fun, but not joy; acquaintances, but not friends; servants, but not faithfulness; leisure, but not peace. You can have the husk of everything for money, but not the kernel.
— Arne Garborg
I have recently had the opportunity to contribute to The Objects that Power the Global Economy, a book by Quartz.
The book is about objects that have impact on our world and our lives. "Each chapter of this book examines an object that is driving radical change in the global economy: how we communicate, what we eat, the way we spend our money. The stories are told through global reporting, original photography and illustration by award-winning artists, contributions from business visionaries, data visualization, and interactive features." (Quartz).
the posters
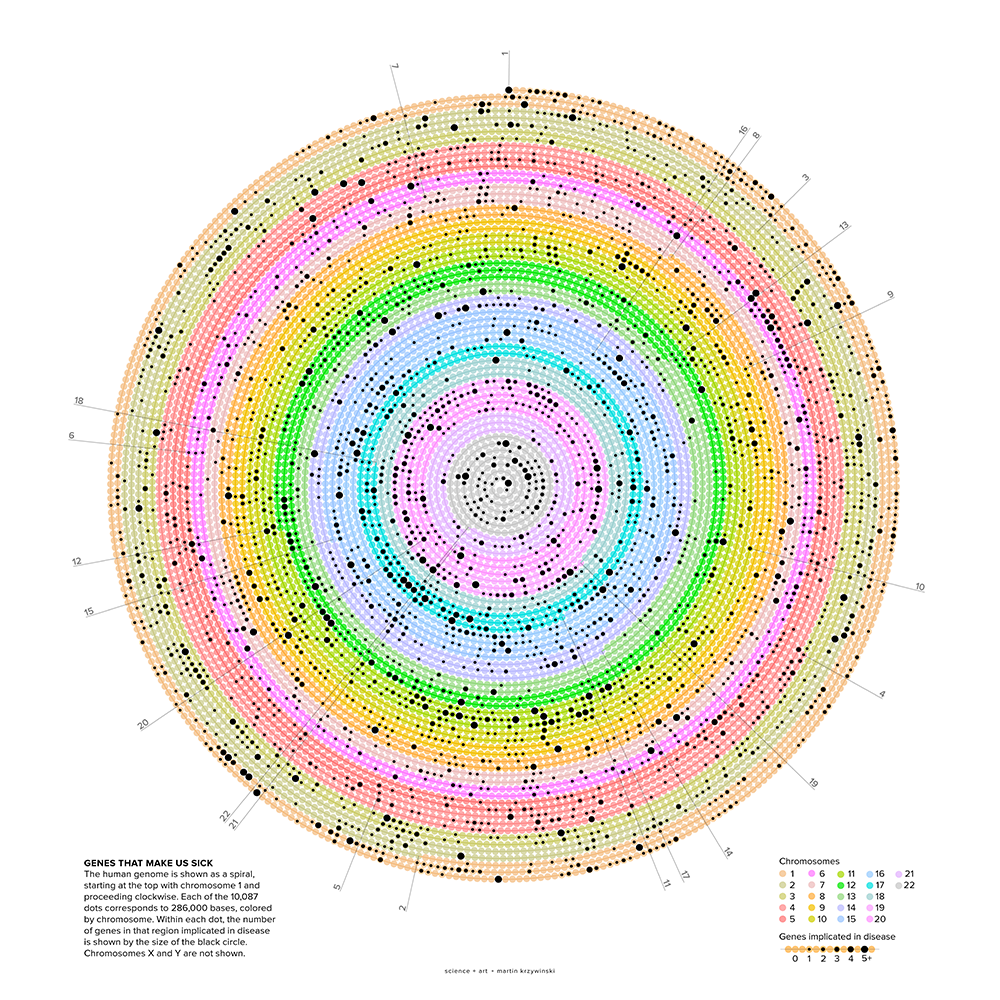
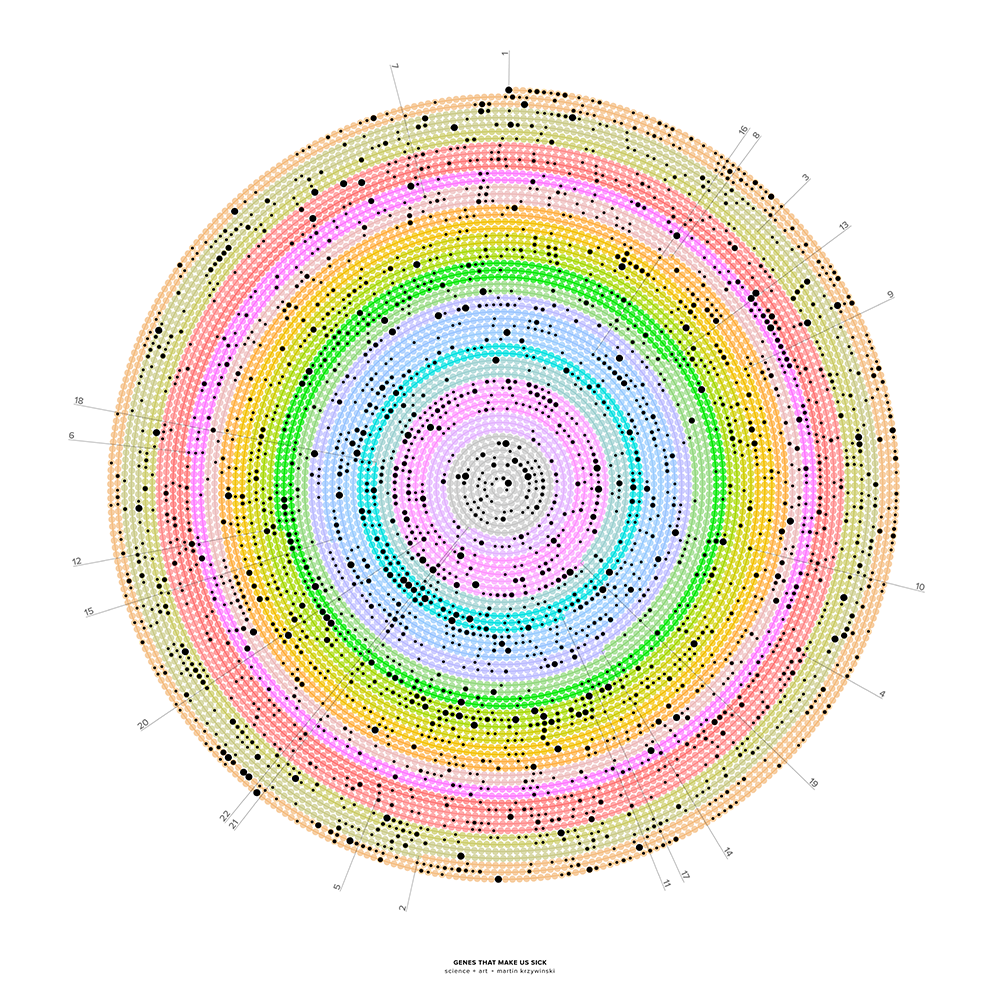
The human genome is shown as a spiral, starting at the top with chromosome 1 and proceeding clockwise. The spiral is formed by 10,087 segments that correspond to 286,000 bases each. Segments that contain genes implicated in disease are indicated by dots, sized by the number of genes. Chromosomes X and Y are not shown.




where disease hides in the genome
My illustration is of the human genome with a focus on the genes that have been implicated in disease.
We have about 30,000 genes and about half of these play some role in disease.
You can peruse what we know about the connection between genetics and illness at the Online Mendelean Inheritance of Man database. For example, a cursory search for "cancer" results in over 3,500 entries.
It's important to realize that these aren't genes that cause disease—its misregulation and mutations in them that are associated with disease (causality is complicated).
the visualization
The illustration shows the genome as a single line, wound in an Archimedean spiral. Chromosomes 1–22 are shown binned into about 10,000 regions along the spiral. Regions that have genes associated with disease are marked with dots—the size of the dot shows how many such genes are found. Each region corresponds to about 286,000 bases.
In about 73% of the 286 kb regions, there are no genes. In about 18%, there is a single gene and in roughly 11% two genes or more.
regions genes
7,321 0
1,812 1
556 2
221 3
85 4
93 5+
Winding the genome up in a spiral creates a compact representation. Squishing a line onto a page can be tricky.
Luckily, space filling curves like the Hilbert curve are very efficient at doing this. I've previously shown the genome along a Hilbert curve for a Scientific American Graphic Science page.
the artwork
In the book, the image is printed on a black background.

Nasa to send our human genome discs to the Moon
We'd like to say a ‘cosmic hello’: mathematics, culture, palaeontology, art and science, and ... human genomes.



Comparing classifier performance with baselines
All animals are equal, but some animals are more equal than others. —George Orwell
This month, we will illustrate the importance of establishing a baseline performance level.
Baselines are typically generated independently for each dataset using very simple models. Their role is to set the minimum level of acceptable performance and help with comparing relative improvements in performance of other models.
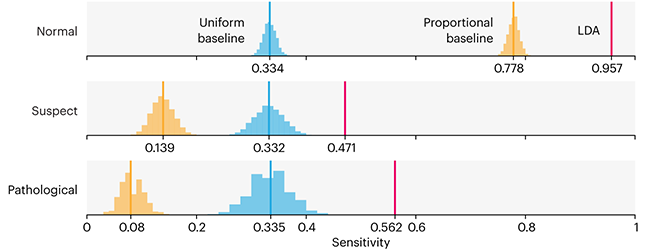
Unfortunately, baselines are often overlooked and, in the presence of a class imbalance5, must be established with care.
Megahed, F.M, Chen, Y-J., Jones-Farmer, A., Rigdon, S.E., Krzywinski, M. & Altman, N. (2024) Points of significance: Comparing classifier performance with baselines. Nat. Methods 20.
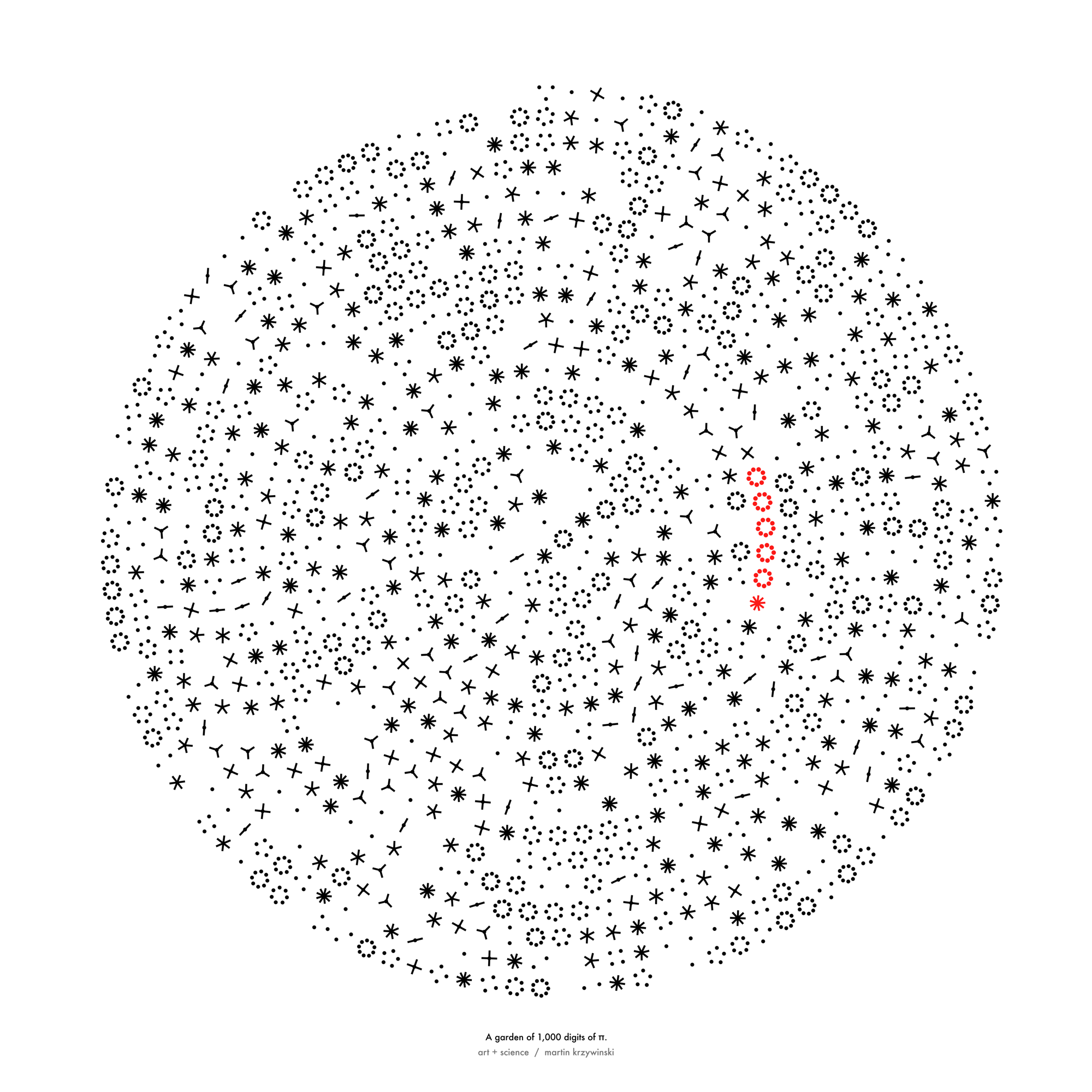
Happy 2024 π Day—
sunflowers ho!
Celebrate π Day (March 14th) and dig into the digit garden. Let's grow something.
How Analyzing Cosmic Nothing Might Explain Everything
Huge empty areas of the universe called voids could help solve the greatest mysteries in the cosmos.
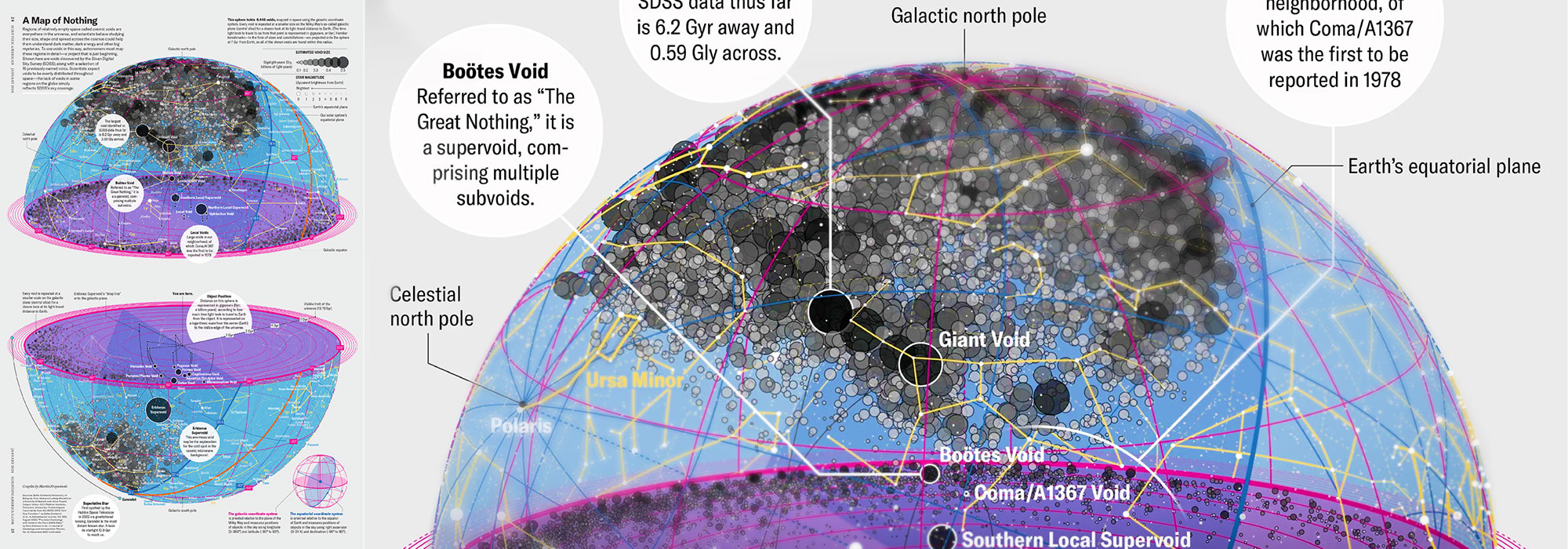
My graphic accompanying How Analyzing Cosmic Nothing Might Explain Everything in the January 2024 issue of Scientific American depicts the entire Universe in a two-page spread — full of nothing.
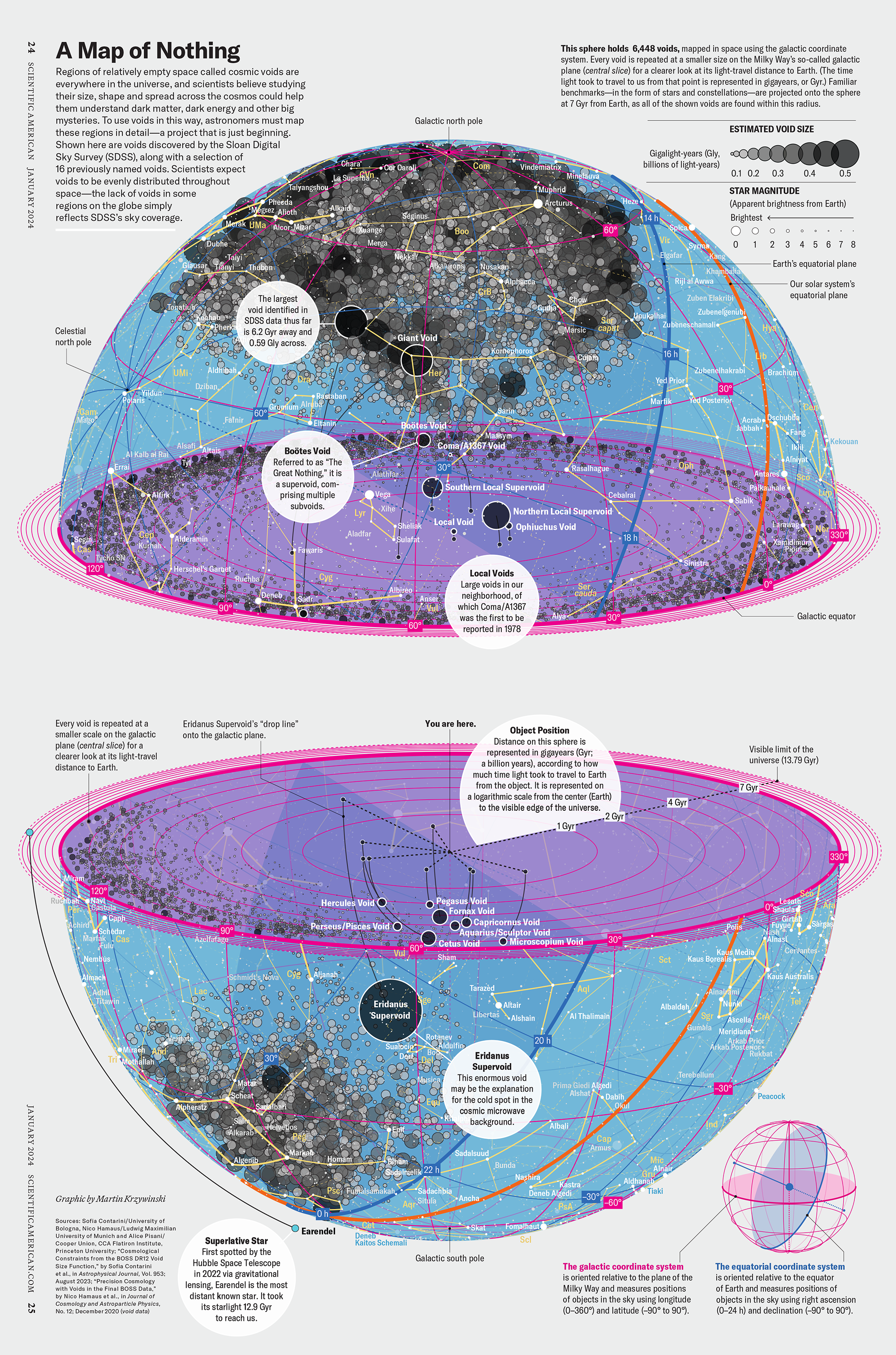
The graphic uses the latest data from SDSS 12 and is an update to my Superclusters and Voids poster.
Michael Lemonick (editor) explains on the graphic:
“Regions of relatively empty space called cosmic voids are everywhere in the universe, and scientists believe studying their size, shape and spread across the cosmos could help them understand dark matter, dark energy and other big mysteries.
To use voids in this way, astronomers must map these regions in detail—a project that is just beginning.
Shown here are voids discovered by the Sloan Digital Sky Survey (SDSS), along with a selection of 16 previously named voids. Scientists expect voids to be evenly distributed throughout space—the lack of voids in some regions on the globe simply reflects SDSS’s sky coverage.”
voids
Sofia Contarini, Alice Pisani, Nico Hamaus, Federico Marulli Lauro Moscardini & Marco Baldi (2023) Cosmological Constraints from the BOSS DR12 Void Size Function Astrophysical Journal 953:46.
Nico Hamaus, Alice Pisani, Jin-Ah Choi, Guilhem Lavaux, Benjamin D. Wandelt & Jochen Weller (2020) Journal of Cosmology and Astroparticle Physics 2020:023.
Sloan Digital Sky Survey Data Release 12
Alan MacRobert (Sky & Telescope), Paulina Rowicka/Martin Krzywinski (revisions & Microscopium)
Hoffleit & Warren Jr. (1991) The Bright Star Catalog, 5th Revised Edition (Preliminary Version).
H0 = 67.4 km/(Mpc·s), Ωm = 0.315, Ωv = 0.685. Planck collaboration Planck 2018 results. VI. Cosmological parameters (2018).
constellation figures
stars
cosmology
Error in predictor variables
It is the mark of an educated mind to rest satisfied with the degree of precision that the nature of the subject admits and not to seek exactness where only an approximation is possible. —Aristotle
In regression, the predictors are (typically) assumed to have known values that are measured without error.
Practically, however, predictors are often measured with error. This has a profound (but predictable) effect on the estimates of relationships among variables – the so-called “error in variables” problem.
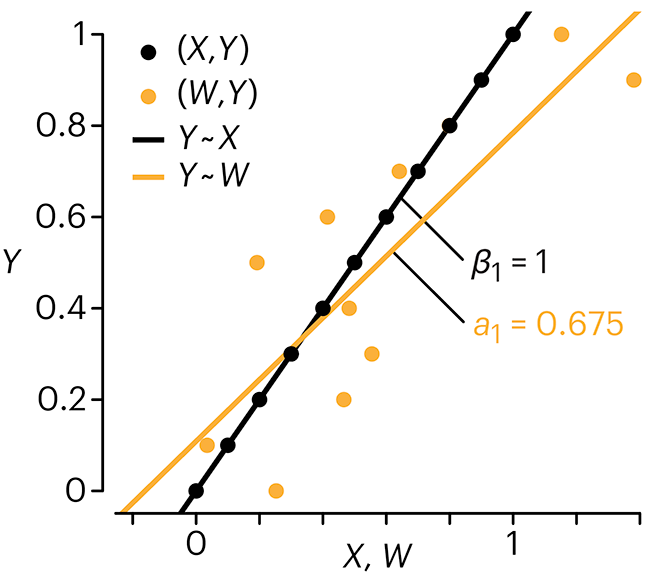
Error in measuring the predictors is often ignored. In this column, we discuss when ignoring this error is harmless and when it can lead to large bias that can leads us to miss important effects.
Altman, N. & Krzywinski, M. (2024) Points of significance: Error in predictor variables. Nat. Methods 20.
Background reading
Altman, N. & Krzywinski, M. (2015) Points of significance: Simple linear regression. Nat. Methods 12:999–1000.
Lever, J., Krzywinski, M. & Altman, N. (2016) Points of significance: Logistic regression. Nat. Methods 13:541–542 (2016).
Das, K., Krzywinski, M. & Altman, N. (2019) Points of significance: Quantile regression. Nat. Methods 16:451–452.
Convolutional neural networks
Nature uses only the longest threads to weave her patterns, so that each small piece of her fabric reveals the organization of the entire tapestry. – Richard Feynman
Following up on our Neural network primer column, this month we explore a different kind of network architecture: a convolutional network.
The convolutional network replaces the hidden layer of a fully connected network (FCN) with one or more filters (a kind of neuron that looks at the input within a narrow window).
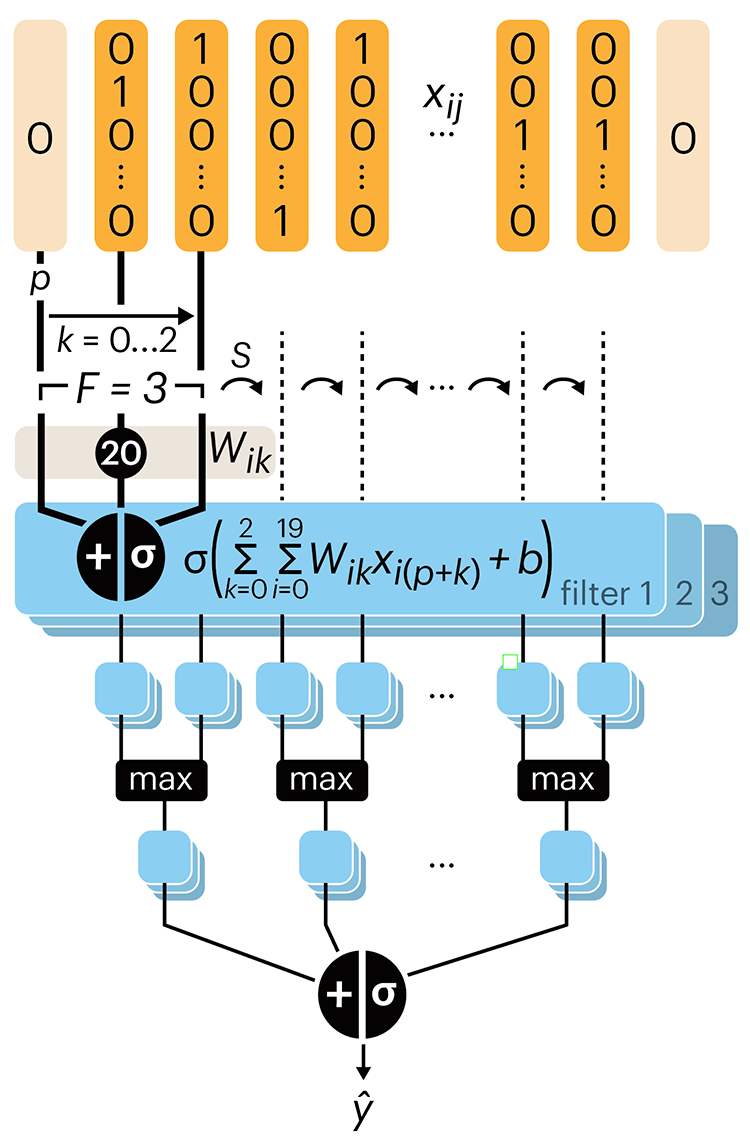
Even through convolutional networks have far fewer neurons that an FCN, they can perform substantially better for certain kinds of problems, such as sequence motif detection.
Derry, A., Krzywinski, M & Altman, N. (2023) Points of significance: Convolutional neural networks. Nature Methods 20:1269–1270.
Background reading
Derry, A., Krzywinski, M. & Altman, N. (2023) Points of significance: Neural network primer. Nature Methods 20:165–167.
Lever, J., Krzywinski, M. & Altman, N. (2016) Points of significance: Logistic regression. Nature Methods 13:541–542.