Learning Circos
Bioinformatics and Genome Analysis
Institut Pasteur Tunis Tunisia, December 10 – December 11, 2018
v2.00 6 Dec 2018 Download PDF slides
v2.00 6 Dec 2018
A 2- or 4-day practical mini-course in Circos, command-line parsing and scripting. This material is part of the Bioinformatics and Genome Analysis course held at the Institut Pasteur Tunis.
Quick links
BCGA 2018 | 1-day Circos course | Circos documentation best practices getting started | Brewer palette swatches | Color resources | Nature Methods Points of View Points of Significance
Visualizing and parsing .maf alignments
Additional material — Day 4
9h00 - 10h30 | Lecture 1 — Perl data structure and regular expression refresher
11h00 - 12h30 | Lecture (practical) 2 — Parsing .maf Ebola strain alignments on the command line
14h00 - 15h30 | Lecture (practical) 3 — Parsing .maf Ebola strain alignments with Perl
16h00 - 18h00 | Lecture (practical) 4 — Visualizing Ebola strain alignments
Concepts covered
.maf multiple-alignment data format, intermediate scripting, regular expressions, flexible parsing with Perl, creating Circos input data files, visualizing alignments between large number of sequences
Parsing .maf Ebola strain alignments on the command line
We will download and parse .maf multi-way alignments in between Ebola strains. To learn more about differences in Ebola strains, see Ebolavirus comparative genomics by Jun et al.
Downloading Ebola Multiz alignments
I have downloaded alignments between about 160 strains of Ebola virus from the UCSC genome browser table viewer. You'll find this file in
ebola.multiz.txt
If you are not familiar with the UCSC genome browser table viewer, you should download the file yourself to familiarize yourself with how the table viewer works.
In the interface, select the following parameters
clade: Viruses
genome: Ebola virus
group: Comparative genomics
track: 160 strains
table: Multiz Align (strainName160way)
format: MAF - multiple alignment format
If you don't define
output file:
then the output will be sent to your browser in plain-text. If you define the file (which you should), you'll browser will download it.
Once you're ready, take a look at the file
> head ebola.multiz.txt
#maf version=1
a score=183000.000000
s eboVir3.KM034562v1 0 1 + 18957 C
s Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 0 1 + 18959 C
i Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 N 0 C 0
s Guinea_Gueckedou-C07_2014.Guinea_Gueckedou-C07_2014 0 1 + 18959 C
i Guinea_Gueckedou-C07_2014.Guinea_Gueckedou-C07_2014 N 0 C 0
s Guinea_Gueckedou-C05_2014.Guinea_Gueckedou-C05_2014 0 1 + 18959 C
i Guinea_Gueckedou-C05_2014.Guinea_Gueckedou-C05_2014 N 0 C 0
...
Details about the MAF format can be found here
http://www.bx.psu.edu/~dcking/man/maf.xhtml
Basically, each alignment is represented by a series of lines. Alignments are separated by a blank line.
The first line in the alignment is the score
a score=1230231.000000
which we're not going to worry about for today. The next series of lines relate the start and length of the alignment in each genome. For each alignment there will be a long list of alignments (we have 160 Ebola strains). For now, you only need to pay attention to the lines that start with s, which give the genome, start and length of the alignment. For example
s Zaire_1995.Zaire_1995 18955 2 + 18961 GT
s Bundibugyo_2007.Bundibugyo_2007 18935 2 + 18940 GT
s Reston_1996.Reston_1996 18885 2 + 18890 GT
Tell us that the region [18955,18956] in strain Zaire_1995 aligns to region [18935,18936] in strain Bundibugyo_2007.
Our goal will be to take this file and parse it into a format that Circos understands. We'll use the link format
CHR1 START1 END1 CHR2 START2 END2
since we'll want to draw the alignments as links.
We'll choose a reference strain Zaire_1995 and report alignments between this strain and all other strains as separate lines. For example, something like
Zaire_1995 18955 18956 Bundibugyo_2007 18935 18936 offset=20
where the offset variable is an option that stores the difference between start positions in the two strains.
Quick parsing on the command-line
We'll start by doing as much as possible on the command-line.
Let's pull out alignments between Zaire_1995 and Reston_1996.
For each multi-line alignment block we need to pull out the lines for the two strains. This can be a little annoying because the alignment record is multi-line and most of the command-line tools apply to one line at a time.
Luckily, we can use awk and change the record separator RS to a double new line. With this set, awk will iterate over alignment blocks, which will be treated as single strings with newlines separating the individual lines in the alignment.
We can then prefix each line in each record by the record number NR.
#parsemaf.sh
cat ../1/ebola.multiz.txt | grep -v \# | \
awk 'BEGIN {RS="\n\n"} {print gensub(/^|\n/,""sprintf("\n%04d",NR)" ","g")}'
For more details about awk's gensub() see
https://www.gnu.org/software/gawk/manual/html_node/String-Functions.html
Here is our parsed file. Now each alignment has a unique number.
# ebola.multiz.numbered.txt
0001 a score=183000.000000
0001 s eboVir3.KM034562v1 0 1 + 18957 C
0001 s Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 0 1 + 18959 C
0001 i Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 N 0 C 0
...
0002 a score=201600.000000
0002 s eboVir3.KM034562v1 1 1 + 18957 G
0002 s Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 1 1 + 18959 G
0002 i Guinea_Kissidougou-C15_2014.Guinea_Kissidougou-C15_2014 C 0 C 0
...
We can now work with this file using grep and join. For each alignment, we pull out the score and lines for Zaire and Reston into separate files.
# parsemaf.sh
for var in score Zaire_1995 Reston_1996 ; do
grep $var ebola.multiz.numbered.txt | sort > tmp.$var.txt
done
Now we can join the lines on the unique alignment record number.
# parsemaf.sh
join tmp.score.txt tmp.Zaire_1995.txt | join - tmp.Reston_1996 > tmp.z.vs.r.txt
There are more columns here than what we want, such as the genome size. Let's pull out only the strain name, start, length and score. We'll also remove all non-space characters after a period. They're useless to us here, since they're either trailing zeros in the alignment score or a repetition of the strain name.
# parsemaf.sh
cut -d " " -f 3,5-7,12-14 tmp.z.vs.r.txt | sed 's/[.][^ ]*//gi' > tmp.z.vs.r.2.txt
Keep in mind that the set of regular expressions supported by different command-line tools can be different. For example Perl's character class for a digit \d is not supported in grep, which uses "[:digit:]". For a complete list of grep and sed character classes see
grep character classes
https://www.gnu.org/software/grep/manual/html_node/Character-Classes-and-Bracket-Expressions.html
sed character classes
https://www.gnu.org/software/sed/manual/html_node/Character-Classes-and-Bracket-Expressions.html
perl character classes
https://perldoc.perl.org/perlrecharclass.html
Finally, we'll go back to awk to reorder the fields, add length to start to get the end position and report the offset and score as options that Circos can read.
# parsemaf.sh
cat tmp.z.vs.r.2.txt | awk '{ print $2,$3,$3+$4,$5,$6,$6+$7,"offset="$6-$3","$1 }' > z.vs.r.txt
# z.vs.r.txt
Zaire_1995 1 2 Reston_1996 1 2 offset=0,score=201600
Zaire_1995 2 3 Reston_1996 2 3 offset=0,score=221100
Zaire_1995 3 4 Reston_1996 3 4 offset=0,score=207298
Zaire_1995 4 5 Reston_1996 4 5 offset=0,score=248500
...
Notice that I wrote to temporary files in the current directory in the script and prefixed them with tmp. Personally, I prefer this to a .tmp extension since my terminal settings list files in alphabetical order. So this way, the temporary files are listed together.
At the end of the script you'll see the files being deleted
# parsemaf.sh
\rm -f tmp*
You can always remove this step if you want to see the intermediate steps.
Theoretically, we could pipe all the steps together but breaking them down into individual commands makes debugging easier.
We now have a Circos link file that we can visualize and all we need is the karyotype file. Let's just make it by hand (we'll automate this step in the next step).
We can get the genome size of the strains by reading it off from the alignment file (it's the column after the strand).
# karyotype.txt
chr - Zaire_1995 zaire 0 18961 black
chr - Reston_1996 reston 0 18887 black
#!/bin/bash
# Insert alignment record number in front of each line in the alignment
cat ../1/ebola.multiz.txt | grep -v \# | \
awk 'BEGIN {RS="\n\n"} {print gensub(/^|\n/,""sprintf("\n%04d",NR)" ","g")}' > ebola.multiz.numbered.txt
# Create temporary files with score and Zaire and Reston lines from
# each alignment, sorted by the alignment record number. Do not
# report the "i" lines
for var in score Zaire_1995 Reston_1996 ; do
grep $var ebola.multiz.numbered.txt | grep -v -w i | sort > tmp.$var.txt
done
# Join the lines on the alignment record number.
join tmp.score.txt tmp.Zaire_1995.txt | join - tmp.Reston_1996.txt > tmp.z.vs.r.txt
# Pull out the relevant columns and remove everything
# after the period.
cut -d " " -f 3,5-7,12-14 tmp.z.vs.r.txt | sed 's/[.][^ ]*//gi' > tmp.z.vs.r.2.txt
# Reformat
cat tmp.z.vs.r.2.txt | awk '{ print $2,$3,$3+$4,$5,$6,$6+$7,"offset="$6-$3","$1 }' > z.vs.r.txt
# Remove tmp files
\rm -f tmp*
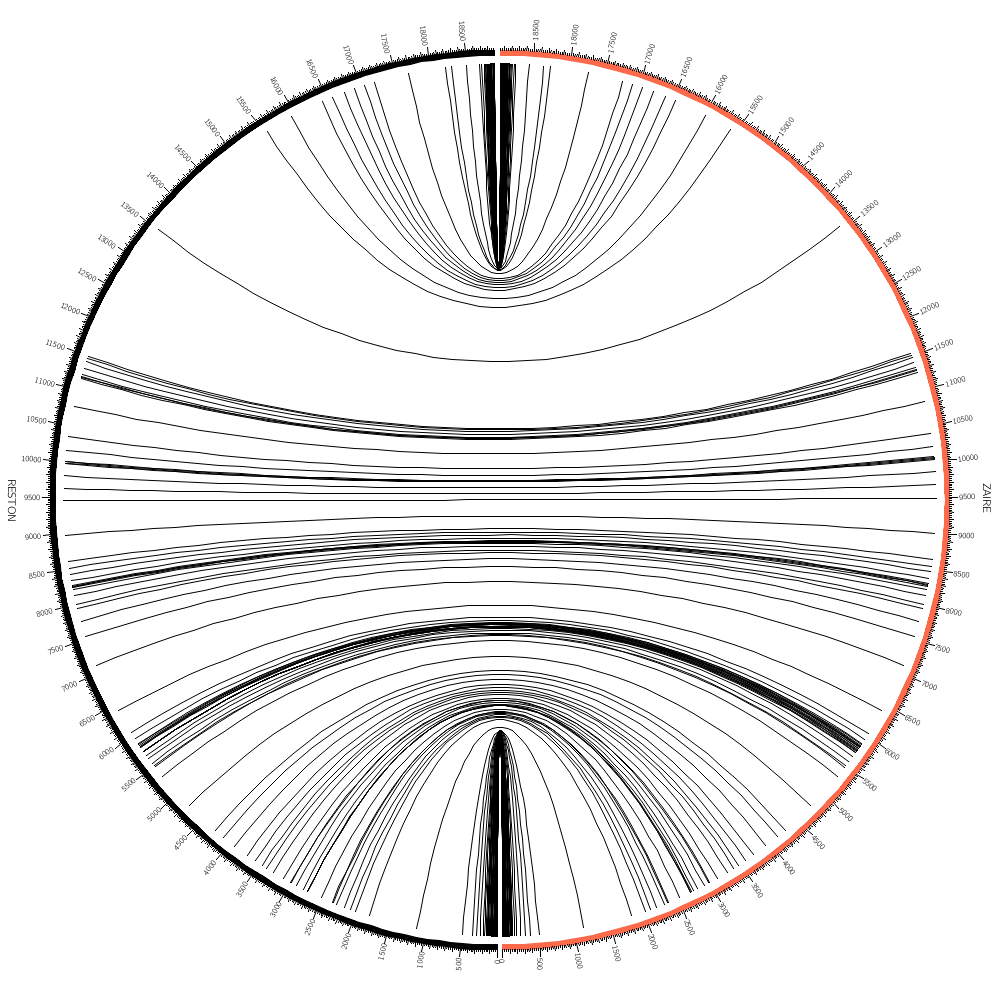
These kinds of plots are vernacularly called "Jupiter plots". I'm sure you can guess why.
karyotype = ../2/karyotype.txt
We have parsed alignments for Zaire_1995 vs Reston_1996 Let's make the Zaire strain segment red.
chromosomes_color = /Zaire/:red
Comment out the line below and see what happens to the image. Can you figure out what's going on?
chromosomes_reverse = /Zaire/
Now we plot the alignments between the two strains.
<links>
<link>
file = ../2/z.vs.r.txt
radius = 0.98r
bezier_radius = 0r
color = black
thickness = 1
</link>
</links>