Learning Circos
Bioinformatics and Genome Analysis
Institut Pasteur Tunis Tunisia, December 10 – December 11, 2018
v2.00 6 Dec 2018 Download PDF slides
v2.00 6 Dec 2018
A 2- or 4-day practical mini-course in Circos, command-line parsing and scripting. This material is part of the Bioinformatics and Genome Analysis course held at the Institut Pasteur Tunis.
Quick links
BCGA 2018 | 1-day Circos course | Circos documentation best practices getting started | Brewer palette swatches | Color resources | Nature Methods Points of View Points of Significance
Visualizing and Parsing clustal alignments
Additional material — Day 3
9h00 - 10h30 | Lecture 1 — Visualization strategies
11h00 - 12h30 | Lecture (practical) 2 — Parsing clustal alignments on command line
14h00 - 15h30 | Lecture (practical) 3 — Parsing clustal alignments with Perl
16h00 - 18h00 | Lecture (practical) 4 — Plotting clustal alignments
Concepts covered
designing effective visualizations, parsing clustal alignments, advanced Perl techniques, using Circos tools
Parsing clustal alignments on command line
In this lecture you'll use the Circos tool clustal2link to parse clustal multi alignment files. You'll also create Circos images that show the alignments. You will also learn how to parse clustal using command-line tools.
You'll find clustal alignments in
day.3/data/alignment.aln
The output format is very easy to understand and is human-readable, which doesn't always make it easily computer-readable!
http://meme-suite.org/doc/clustalw-format.html
One of the Circos tools is clustal2link, which parses clustal alignments and generates Circos input files to draw the alignments and coverage. The tools/ directory in the Circos installation consist of the following
binlinks/ generate histogram tracks of link counts and sizes from link files
bundlelinks/ combine multiple links together based on distance and size
calcdatarange/ determine which regions of the ideogram there is data, useful for cropping
categoryviewer/ early version of the tableviewer (see below)
clustal2link/ parse clustal alignments and visualize them and their coverage
colorinterpolate/ interpolate colors in various color spaces, useful for making color ramps
connectogram/ parse and draw neural connectogram images
convertlinks/ convert link files from 2-line format to 1-line format (deprecated)
filterlinks/ filter links from a file based on position and size
matrix/ convert a list of cell values into a matrix and vice versa
orderchr/ generate an ideogram order that minimizes the number of crossed links
randomdata/ generate random data with given properties, useful for debugging
randomlinks/ generate random links with given properties, useful for debugging
resample/ downsample a track data file
tableviewer/ generate a Circos image of tabular data, http://mkweb.bcgsc.ca/tableviewer
You'll find this tool in the Circos installation directory under
tools/clustal2link
relative to the Circos installation directory.
As most of the tools, clustal2link has two components. First, a script and configuration file
bin/clustal2link
etc/clustal2link.conf
that parse the clustal files. By default, these generate data in
circos/data
And second, the Circos configuration files in
circos/etc
These configuration files are design to make use of parsed files.
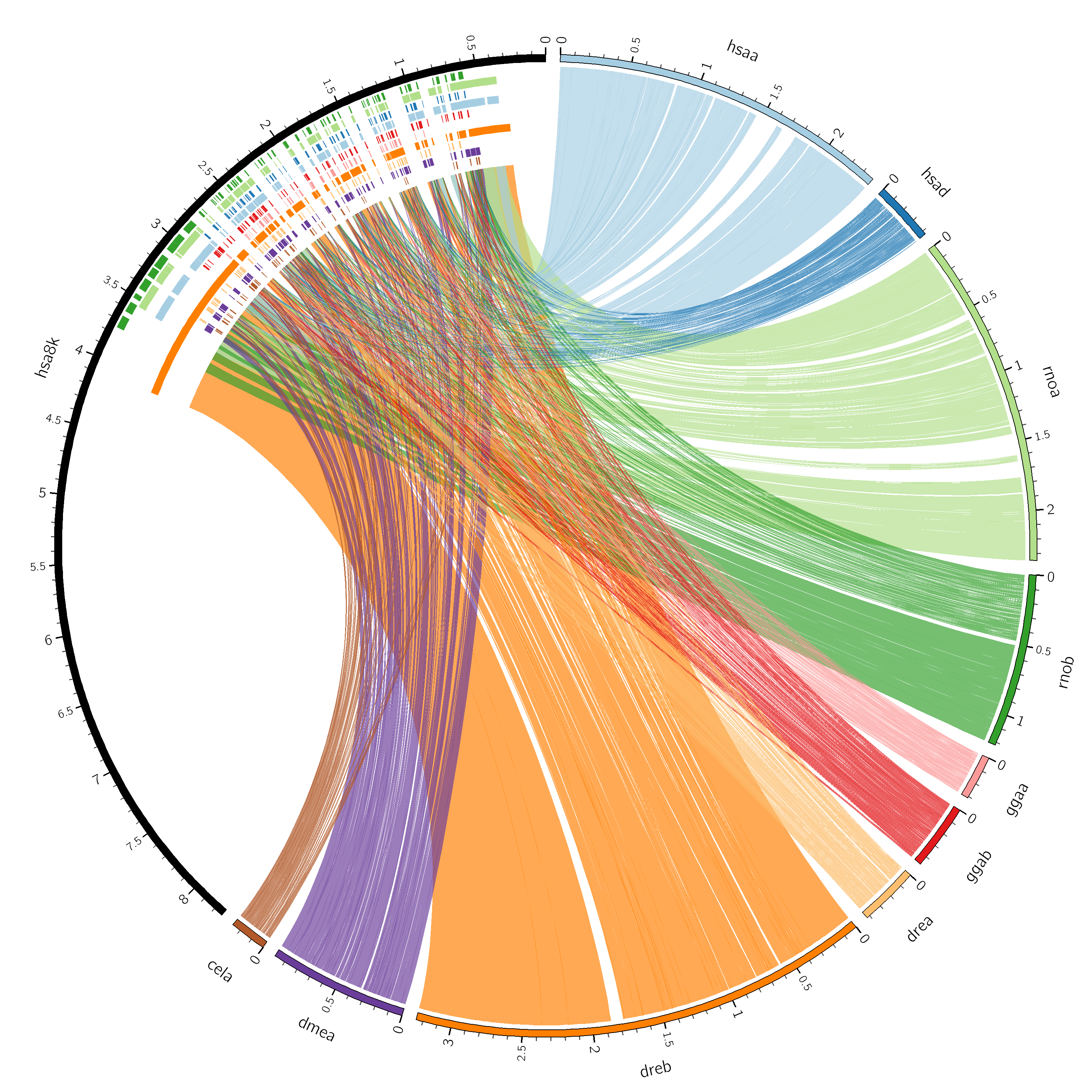
Let's quickly run clustal2link it on the data and see what the image looks like.
#parse the alignment files
> ./run.sh
Take a look at the output in
#data/
-rw-r--r-- 1 martink users 742 Oct 17 10:41 highlight.celA.txt
-rw-r--r-- 1 martink users 1121 Oct 17 10:41 highlight.dmeA.txt
-rw-r--r-- 1 martink users 1236 Oct 17 10:41 highlight.dreA.txt
-rw-r--r-- 1 martink users 768 Oct 17 10:41 highlight.dreB.txt
-rw-r--r-- 1 martink users 894 Oct 17 10:41 highlight.ggaA.txt
-rw-r--r-- 1 martink users 1212 Oct 17 10:41 highlight.ggaB.txt
-rw-r--r-- 1 martink users 975 Oct 17 10:41 highlight.hsaA.txt
-rw-r--r-- 1 martink users 1036 Oct 17 10:41 highlight.hsaD.txt
-rw-r--r-- 1 martink users 1003 Oct 17 10:41 highlight.rnoA.txt
-rw-r--r-- 1 martink users 1136 Oct 17 10:41 highlight.rnoB.txt
-rw-r--r-- 1 martink users 305 Oct 17 10:41 karyotype.txt
-rw-r--r-- 1 martink users 18429 Oct 17 10:41 links.txt
The format of these files should already be familiar to you.
Now run Circos and look at the image
>circos
#!/bin/bash
# This is the directory where the Circos tools are installed
# on my computer at work. But for your setup, it'll be different.
# Change this to the right directory. Take a look in /root/modules/circos
# to find it.
CTOOLS=/home/martink/work/circos/svn/tools/
mkdir data
$CTOOLS/clustal2link/bin/clustal2link -aln ../../data/alignment.aln -idref hsa8K -dir data -debug
Sample Circos configuration for clustal multiple alignments.
This is a pretty complicated configuration that uses many advanced features. You'll build up your own image from scratch today. Use this file as a reference.
The data files required are generated by tools/clustal2link/
highlight.*.txt
links.txt
karyotype.txt
In addition to these files, you need to provide the following
sequences.conf - order of sequences for highlight tracks (all sequences except for reference)
seq.colors.conf - RGB colors for each sequence
List of sequence IDs to use as ideogram names
<<include sequences.conf>>
<colors>
Append colors for each sequence ID to color definitions.
<<include seq.colors.conf>>
</colors>
karyotype = karyotype.txt
chromosomes_units = 1000
chromosomes_display_default = yes
You can set this (and any other parameter) at command line
> circos -param refseq=hsa8K
refseq = hsa8K
reverse everything, except ID that matches /8/ (hsa8K).
chromosomes_reverse = /./,-/conf(refseq)/
order of chromosomes.
chromosomes_order = hsa8K,celA,dmeA,dreB,dreA,ggaB,ggaA,rnoB,rnoA,hsaD,hsaA
make hsa8K (e.g. reference sequence) to occupy 50% of the circle chromosomes_scale = conf(refseq):0.5r make segments of all sequences equal size chromosomes_scale = /./=1rn crop out regions of the reference sequence chromosomes_breaks = -hsa8K:4.2-7.8
<links>
<link>
ribbon = yes
file = data/links.txt
radius = 0.99r
color = black
bezier_radius = 0r
crest = 0.2
<rules>
<rule>
This rule colors each link by its originating sequence color and sets the z-value (depth) of the link to be proportional to negative of originating size. Small links (larger z-value) will therefore be drawn on top of larger links (smaller z-value)
condition = 1
color = eval(sprintf("%s_a2",lc var(chr1)))
radius2 = 0.79r
z = eval(-var(size1))
</rule>
</rules>
</link>
</links>
initial radius, width and padding of highlight tracks.
h_0 = 0.80
h_width = 0.015
h_pad = 0.005
<plots>
We can automate drawing tracks by dynamically defining the position and file name of each track based on a counter variable that Circos uses for each track. This allows us to have an identical definition for tracks that will be dynamically parsed at runtime.
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
<<include highlight.conf>>
</plots>
Let's get started writing a script to parse the clustal alignments.
Although you can already use clustal2link, often you'll need to parse your own files. You might be surprised that about half of the questions about Circos that I get are not about how to draw the image but how to parse files!
In this lecture, you'll use command-line tools to parse the alignment files. In the next lecture, we'll look at how we can use Perl to build a more robust parser.
Because parsing files on the command-line is a very important skill to develop, I've dedicated a lot of time to this part. Make sure that you fully understand everything in this section before proceeding. This example will be a little more advanced than the Ebola strain alignments from the last lecture.
Run the bash script that generates Circos link files from the alignment
>./parseclustal.sh
The output is ready in
parsed/
You'll notice that I chose not to generate the output to the current directory. It's tidier to keep it separate.
The first thing the script does is create a file of unique sequence ids.
FILE=../../data/alignment.aln
cat $FILE | cut -d " " -f 1 | sort -u | grep -v -i clustal | grep "[a-z]" > $OUTDIR/id.txt
By now you should be pretty familiar with what is being done here. We list the file, keep only the first column (space as delimiter), create a list of unique column values with sort -u and then remove the line that contains the "CLUSTAL" header and finally only keep lines with letters in them (some were blank in the original alignment).
> cat id.txt
celA
dmeA
dreA
dreB
ggaA
ggaB
hsa8K
hsaA
hsaD
rnoA
rnoB
Our next step is to pull out the sequence for each sequence id and store it in a file, with one base per line. We do this by looping over all the sequence ids. Here the backticks indicate a command whose output is treated as the list over which to loop.
For example, to loop over a few values entered manually
for fruit in banana apple orange; do
echo "I like $fruit"
done
If you want a list of numbers, use the command-line seq utility which provides a list of numbers up to a value
> seq 3
1
2
3
or between two values
> seq 3 6
3
4
5
6
The following will loop over i = 1..10.
for i in `seq 1 10`; do
echo "number $i"
done
So, to loop over sequence IDs, we cat the contents of id.txt for the loop list
for id in `cat $OUTDIR/id.txt`; do
grep $id $FILE | tr -s " " | cut -d " " -f 2 | fold -c1 > $OUTDIR/$id.seq.txt
done
We're half-way there! We now have a file of the sequence for each id
#hsa8K.seq.id
A
C
C
G
...
If you look at the original alignment file, you'll see that there are a lot of spaces in the file that we don't need. We can deal with these spaces by removing runs of spaces
tr -s " "
but often there are tabs, leading spaces, trailing spaces and empty lines that you want to remove too. For this, I have written the script shrinkwrap.sh which accomplishes this. Basically it assumes that the file has some stray spaces and removes them all, keeping only non-blank lines.
> cat ../../data/alignment/shrinkwrap.sh
CLUSTAL 2.1 multiple sequence alignment
hsaA ------------------------------------------------------------
dreA ------------------------------------------------------------
rnoA ------------------------------------------------------------
dmeA ------------------------------------------------------------
celA ------------------------------------------------------------
hsa8K ACCGGATAATGCCTATCCCAGAAACATTCAAATCAAGCCCATGAGTACCCACATGGCTAA 60
ggaB ------------------------------------------------------------
hsaD ------------------------------------------------------------
...
It also removes any lines that begin with the comment character #. This script is very helpful in cleaning up data files. You can also set the output delimiter if your script expects, for example, a comma-delimited list.
> cat ../../data/alignment/shrinkwrap.sh ,
CLUSTAL,2.1,multiple,sequence,alignment
hsaA,------------------------------------------------------------
dreA,------------------------------------------------------------
rnoA,------------------------------------------------------------
dmeA,------------------------------------------------------------
celA,------------------------------------------------------------
hsa8K,ACCGGATAATGCCTATCCCAGAAACATTCAAATCAAGCCCATGAGTACCCACATGGCTAA,60
ggaB,------------------------------------------------------------
hsaD,------------------------------------------------------------
...
We'll use this script to tidy up our parsing as we go along.
Ok, back to our sequences. You'll see that each sequence length is the same: 9,270 bases.
> wc *seq.txt
9270 9270 18540 celA.seq.txt
9270 9270 18540 dmeA.seq.txt
9270 9270 18540 dreA.seq.txt
9270 9270 18540 dreB.seq.txt
9270 9270 18540 ggaA.seq.txt
9270 9270 18540 ggaB.seq.txt
9270 9270 18540 hsa8K.seq.txt
9270 9270 18540 hsaA.seq.txt
9270 9270 18540 hsaD.seq.txt
9270 9270 18540 rnoA.seq.txt
9270 9270 18540 rnoB.seq.txt
This is the total length of the alignment. We expected the files to have the same length because every sequence is represented at every position along the length of the alignment.
We now use the very useful paste tool, which joins lines from two files into columns. paste is one of those things that you rarely use but when you need it, it's a life saver.
> paste hsa8K.seq.txt celA.seq.txt
A -
C -
C -
G -
...
The first column is from hsa8K and the second from celA. We can also add line numbers.
> paste hsa8K.seq.txt celA.seq.txt | cat -n
1 A -
2 C -
3 C -
4 G -
...
See how stray spaces were introduced in this process? Many command-line tools introduce a default tab delimiter and sometimes have leading spaces. By piping everything through our shrinkwrap.sh script, we can clean all this up.
> paste hsa8K.seq.txt celA.seq.txt | cat -n | ../shrinkwrap.sh
1 A -
2 C -
3 C -
4 G -
...
We create a file like this for each alignment pair. You'll see these files as
align.hsa8K.*.txt
What we now need is a range of regions with bases in both sequences. Each time we have a dash in either sequence we know that there is no alignment there.
We can achieve this by first replacing all bases with X and then counting the length of runs with the same 2nd and 3rd column (skipping over the position).
>cat align.hsa8K.celA.txt | tr ATGC X | uniq -f 1 -c
580 1 X -
9 581 X X
13 590 X -
7 603 X X
41 610 X -
2 651 - -
8 653 X -
...
The first number is the number of lines that we saw with the same 2nd and 3rd columns (e.g. X -). The second number is the original first column from the file, which is the start position of the alignment.
What this step has done is provided the length and start of regions in the alignment file, which can be one of the following
X X # alignment
X - # no alignment to 2nd sequence
- X # no alignment to reference
- - # no alignment to either
We don't care about any lines with a dash, since these are regions without an alignment.
>cat align.hsa8K.celA.txt | tr ATGC X | uniq -f 1 -c | grep -v -
9 581 X X
7 603 X X
5 661 X X
7 804 X X
3 834 X X
...
And here are our alignments!
They're not exactly in the format that we want. What we need is
ID1 START1 END1 ID2 START2 END2
But we have
LENGTH START X X
So we can use awk to generate the right format by simply adding the length-1 to the start.
>cat align.hsa8K.celA.txt | tr ATGC X | uniq -f 1 -c | grep -v - \
awk -v ref=$REF -v id=$id '{print ref,$2,$2+$1-1,id,$2,$2+1-2}'
hsa8K 581 589 celA 581 580
hsa8K 603 609 celA 603 602
hsa8K 661 665 celA 661 660
hsa8K 804 810 celA 804 803
hsa8K 834 836 celA 834 833
hsa8K 1367 1370 celA 1367 1366
...
We can quickly see how many alignments we have per pair
> wc -l links.hsa8K*
48 links.hsa8K.celA.txt
75 links.hsa8K.dmeA.txt
79 links.hsa8K.dreA.txt
66 links.hsa8K.dreB.txt
56 links.hsa8K.ggaA.txt
77 links.hsa8K.ggaB.txt
78 links.hsa8K.hsaA.txt
66 links.hsa8K.hsaD.txt
78 links.hsa8K.rnoA.txt
75 links.hsa8K.rnoB.txt
We can also add up the total length of the alignment for each file and get some statistics about the distribution of alignment lengths. Use the histogram script you'll write in Day 4 Lecture 4? It's time to use it again.
> cat links.hsa8K.celA.txt | awk '{print $3-$2+1}' | ../../../scripts/histogram
1.0000> 0 0.000
1.0000 1.9500 2 0.042 0.042 ***
1.9500 2.9000 6 0.125 0.167 ***********
2.9000 3.8500 7 0.146 0.312 *************
3.8500 4.8000 13 0.271 0.583 *************************
4.8000 5.7500 8 0.167 0.750 ***************
5.7500 6.7000 0 0.000 0.750
6.7000 7.6500 4 0.083 0.833 *******
7.6500 8.6000 1 0.021 0.854 *
8.6000 9.5500 1 0.021 0.875 *
9.5500 10.5000 3 0.062 0.938 *****
10.5000 11.4500 2 0.042 0.979 ***
11.4500 12.4000 0 0.000 0.979
12.4000 13.3500 0 0.000 0.979
13.3500 14.3000 0 0.000 0.979
14.3000 15.2500 0 0.000 0.979
15.2500 16.2000 0 0.000 0.979
16.2000 17.1500 0 0.000 0.979
17.1500 18.1000 0 0.000 0.979
18.1000 19.0500 0 0.000 0.979
19.0500 20.0000 1 0.021 1.000 *
20.0000< 0 0.000
n 48
average 5.08333
sd 3.35307
min 1.00000
max 20.00000
sum 244.00000
We see that most alignments between hsa8K and celA are within 1-10 bases long. So let's explore this region of the histogram in more detail
> cat links.hsa8K.celA.txt | awk '{print $3-$2+1}' | ../../../scripts/histogram -max 10 -binsize 1
1.0000> 0 0.000
1.0000 2.0000 2 0.044 0.044 ***
2.0000 3.0000 6 0.133 0.178 ***********
3.0000 4.0000 7 0.156 0.333 *************
4.0000 5.0000 13 0.289 0.622 *************************
5.0000 6.0000 8 0.178 0.800 ***************
6.0000 7.0000 0 0.000 0.800
7.0000 8.0000 4 0.089 0.889 *******
8.0000 9.0000 1 0.022 0.911 *
9.0000 10.0000 4 0.089 1.000 *******
10.0000< 3 0.062
n 48
average 5.08333
sd 3.35307
min 1.00000
max 20.00000
sum 244.00000
Use the histogram script to explore alignment distributions for the other sequences.
Look how much we've been able to achieve without doing any actual custom programming for this example. By having a toolbox of useful scripts like histogram and shrinkwrap.sh (and I'm sure many that you can think about writing yourself), you can stay as close to the command line as possible when analyzing files.
This is the UNIX way: many small tools combined together to break down a complex task into a series of smaller simpler tasks.
In the next lecture, we'll explore how to write a more robust Perl parser for clustal files.
#!/bin/bash
# extract the list of sequence IDs from the alignment file
REF=hsa8K
OUTDIR=parsed
FILE=../../data/alignment.aln
cat $FILE | cut -d " " -f 1 | sort -u | grep -v -i clustal | grep "[a-z]" > $OUTDIR/id.txt
# for each sequence ID, retrieve the sequence and store it with one base per line
for id in `cat $OUTDIR/id.txt`; do
grep $id $FILE | tr -s " " | cut -d " " -f 2 | fold -c1 > $OUTDIR/$id.seq.txt
done
# paste hsa8K together with each other sequence id
# and remove lines with "-" indicating no alignment
# and make a file of all positions (one per line) at
# which bases are aligned
for id in `cat $OUTDIR/id.txt | grep -v $REF` ; do
# convert tabs to a single space, collapse all spaces and
# remove leading spaces and trailing spaces (done via shrinkwrap.sh script)
paste $OUTDIR/$REF.seq.txt $OUTDIR/$id.seq.txt | cat -n | \
./shrinkwrap.sh > $OUTDIR/align.$REF.$id.txt
# The line below is long but none of the steps are particularly complicated
# 1. replace all bases with an X (tr)
# 2. extract runs of lines that have the same fields (uniq), but skipping first field (-f 1)
# and counting the number of runs (-c)
# 3. remove all lines that have gaps (grep)
# 4. write the output to a file but also to STDOUT (tee)
# 5. add the name of the sequence ids to the file and compute start and end of alignment
# based on the start of the alignment and length of runs from uniq
cat $OUTDIR/align.$REF.$id.txt | tr ATGC X | uniq -f 1 -c | \
grep -v - | tee $OUTDIR/runs.$REF.$id.txt | \
awk -v ref=$REF -v id=$id '{print ref,$2,$2+$1-1,id,$2,$2+1-2}' > $OUTDIR/links.$REF.$id.txt
done
#!/bin/bash
# default delimiter is a space
#
# cat file.txt | shrinkwrap.sh
#
# but you can set it by passing an argument
#
# cat file.txt | shrinkwrap.sh ,
# Assign the first argument to delimiter unless it's not defined
# in which case assign just a space
#
# VAR=${1:-DEFAULTVALUE}
#
DELIM=${1:- }
# all spaces to tabs
expand -t1 | \
# collapse all spaces
tr -s " " | \
# only keep lines that have a non-space character
grep '[^ ]' | \
# remove leading and trailing spaces
sed 's/^[ ]//g' | sed 's/[ ]$//g' | \
# remove lines that begin with comment field
grep -v -e '^\#' |
# replace the space with our delimiter string (space by default)
tr " " "$DELIM"